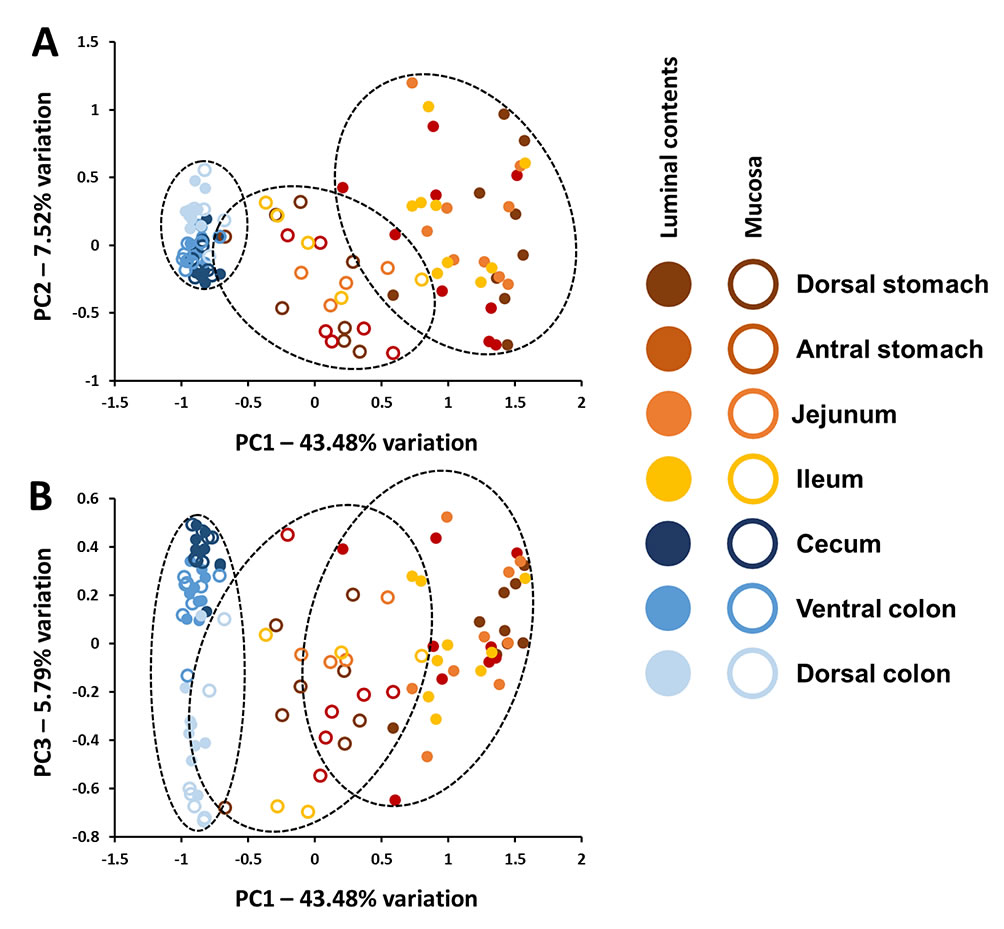
Horses are exquisitely sensitive to non-specific gastrointestinal disturbances as well as systemic and extraintestinal conditions related to gut health, yet minimal data are available regarding the composition of the microbiota present in the equine stomach, small intestine, and cecum and their relation to fecal microbiota. Moreover, there is minimal information regarding the concordance of the luminal and mucosal microbial communities throughout the equine gut. Illumina-based 16S rRNA gene amplicon sequencing of the luminal and mucosal microbiota present in seven regions of the gastrointestinal tract of nine healthy adult horses revealed a distinct compositional divide between the small and large intestines. This disparity in composition was more pronounced within the luminal contents, but was also detected within mucosal populations. Moreover, the uniformity of the gut microbiota was much higher in the cecum and colon relative to that in the stomach, jejunum and ileum, despite a significantly higher number of unique sequences detected in the colon. Collectively, the current data suggest that while colonic samples (a proxy for feces) may provide a reasonable profile of the luminal contents of the healthy equine large intestine, they are not informative with regard to the contents of the stomach or small intestine. In contrast to the distinct difference between the highly variable upper gastrointestinal tract microbiota and relatively uniform large bowel microbiota present within the lumen, these data also demonstrate a regional continuity present in mucosal microbial communities throughout the length of the equine gut.
See full study
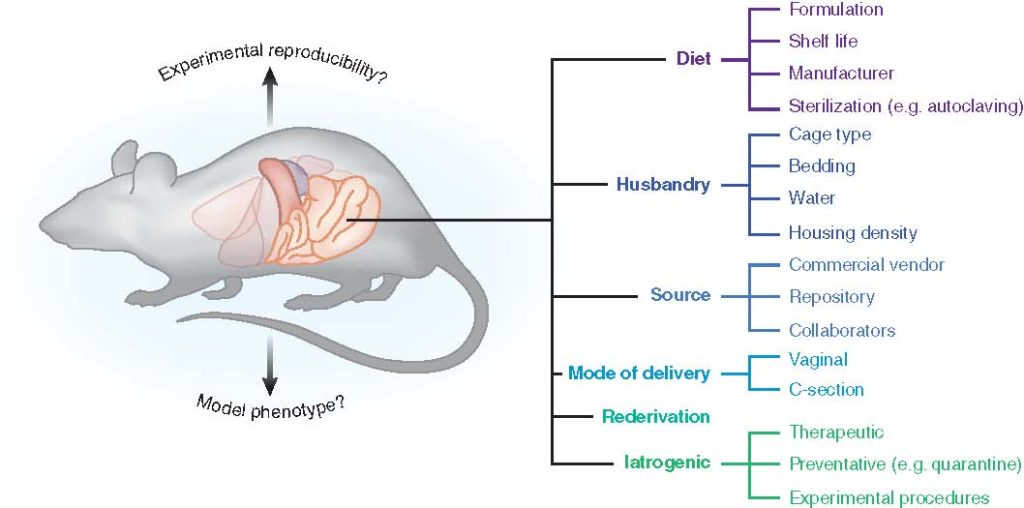
The gut microbiota (GM) plays a critical role in human health and disease. Likewise, it is becoming increasingly evident that changes or disruptions to the GM can have significant effects on animal models and their expressed phenotypes, adding a complex and important variable into basic research and preclinical studies. In this article, we review some of the most common sources of GM variability in rodent models, and discuss measures to address this variability for improved reproducibility.
See full study
Several associations have been made between characteristics of the resident gut microbiota and human health and disease susceptibility. Animal models provide the means to test these correlations prospectively and evaluate causality. Experimental fecal microbiota transfer (FMT), or the intentional transplantation of gut microbes into recipient mice depleted of their autochthonous microbes with antibiotics, is a commonly used method of testing these relationships. The true completeness of microbial transfer through such procedures is poorly documented in the literature, particularly in the context of reciprocal transfer of microbes between recipient and donor mice harboring microbial populations of differing richness and diversity. Moreover, it is unclear whether the use of frozen fecal contents or cecal contents would confer any difference in the outcomes of transfer. Herein, groups of mice colonized with distinct gut microbiota of differing richness and composition were used in a reciprocal FMT study, with different groups receiving transfer of material prepared from fresh cecal contents, fresh feces, or frozen feces. Targeted 16S rRNA gene amplicon sequencing was used at intervals throughout the study to characterize the microbiota. Notably, despite comparable depletion of the microbiota in recipient mice prior to transfer, donor-specific taxa reliably colonized recipients only when relatively rich donor material was transferred to mice originally colonized with a simpler microbiota. It is unclear whether these differences were due to differences in the endogenous recipient microbiota or host factors induced in early life by microbial factors. These findings are of practical import for researchers using FMT to prospectively assess the influence of the gut microbiota in mouse models, and to those studying host-microbial interactions and their influence on gut barrier function.
Vientós-Plotts, A.I., Ericsson, A.C., Rindt, H., Grobman, M.E., Graham, A., Bishop, K., Cohn, L., and C.R. Reinero. (2017) Dynamic changes of the healthy feline airway microbiota. PLoS One, 12(3): e0173818.
See full study
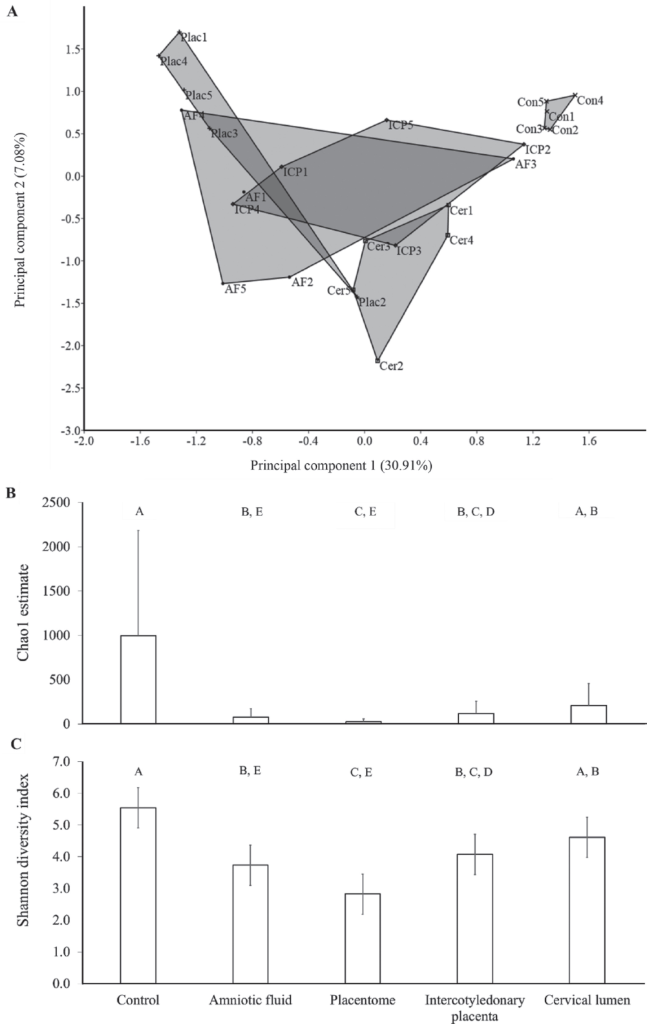
We tested the hypothesis that the uterus of virgin heifers and pregnant cows possessed a resident microbiome by 16S rRNA gene sequencing of the virgin and pregnant bovine uterus. The endometrium of 10 virgin heifers in estrus and the amniotic fluid, placentome, intercotyledonary placenta, cervical lumen, and external cervix surface (control) of 5 pregnant cows were sampled using aseptic techniques. The DNA was extracted, the V4 hypervariable region of the 16S rRNA gene was amplified, and amplicons were sequenced using Illumina MiSeq technology (Illumina Inc., San Diego, CA). Operational taxonomic units (OTU) were generated from the sequences using Qiime v1.8 software, and taxonomy was assigned using the Greengenes database. The effect of tissue on the microbial composition within the pregnant uterus was tested using univariate (mixed model) and multivariate (permutational multivariate ANOVA) procedures. Amplicons of 16S rRNA gene were generated in all samples, supporting the contention that the uterus of virgin heifers and pregnant cows contained a microbiome. On average, 53, 199, 380, 382, 525, and 13,589 reads annotated as 16, 35, 43, 63, 48, and 176 OTU in the placentome, virgin endometrium, amniotic fluid, cervical lumen, intercotyledonary placenta, and external surface of the cervix, respectively, were generated. The 3 most abundant phyla in the uterus of the virgin heifers and pregnant cows were Firmicutes, Bacteroidetes, and Proteobacteria, and they accounted for approximately 40, 35, and 10% of the sequences, respectively. Phyla abundance was similar between the tissues of the pregnant uterus. Principal component analysis, one-way PERMANOVA analysis of the Bray-Curtis similarity index, and mixed model analysis of the Shannon diversity index and Chao1 index demonstrated that the microbiome of the control tissue (external surface of the cervix) was significantly different from that of the amniotic fluid, intercotyledonary placenta, and placentome tissues. Interestingly, many bacterial species associated with postpartum uterine disease (i.e., Trueperella spp., Acinetobacter spp., Fusobacteria spp., Proteus spp., Prevotella spp., and Peptostreptococcus spp.) were also present in the uterus of virgin heifers and of pregnant cows. The presence of 16S rRNA gene sequence reads in the samples from the current study suggests that the uterine microbiome is established by the time a female reaches reproductive maturity, and that pregnancies are established and maintained in the presence of a uterine microbiome.
See full study
See full study
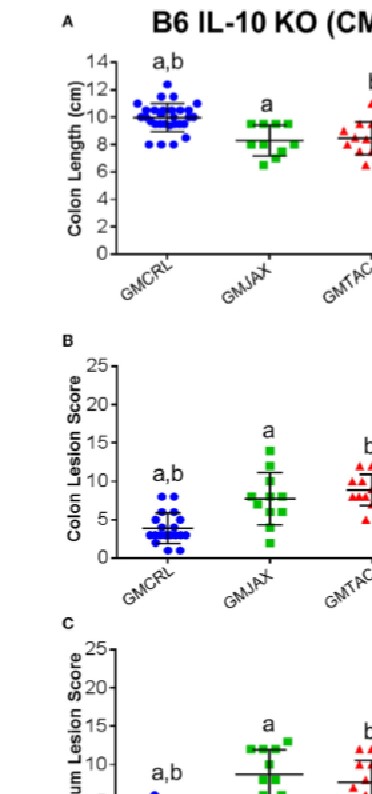
It is estimated that 1.4 million people in the United States suffer from Inflammatory Bowel Disease (IBD), with an overall annual health care cost of more than $1.7 billion. Although the exact etiology of this disease remains unknown, research suggests that it is a multifactorial disease associated with aberrant gastrointestinal microbial populations (dysbiosis). The C57BL/6 and C3H/HeJBir mouse strains with targeted mutations in the IL-10 gene are commonly used models to study IBD. However, anecdotally, disease phenotype can vary in severity from lab to lab. Moreover, studies using germfree and monocolonized mice have suggested that gut microbiota (GM) are critical to disease induction in these models. With recent studies suggesting variation in naturally occurring GM composition and complexity among mouse producers, we hypothesized that differences in these naturally occurring complex GM profiles may modulate disease severity in the IL-10-/- mouse model. To test this hypothesis, we use a technique referred to as complex microbiota targeted rederivation (CMTR) to transfer genetically identical C57BL/6 IL-10-/- and C3H/HeJBir IL-10-/- embryos into surrogate CD-1 or C57BL/6 dams from different commercial producers with varying microbiota complexity and composition. We found that disease severity significantly and reproducibly differed among mice in both IL-10-/- strains, dependent on differing maternally inherited GM. Furthermore, disease severity was associated with alterations in relative abundance of several physiologically relevant bacterial species. These findings suggest that the composition of the resident GM is a primary determinant of disease severity in IBD and provide proof-of-concept that CMTR can be used to investigate the contribution of contemporary complex GM on disease phenotype and reproducibility.
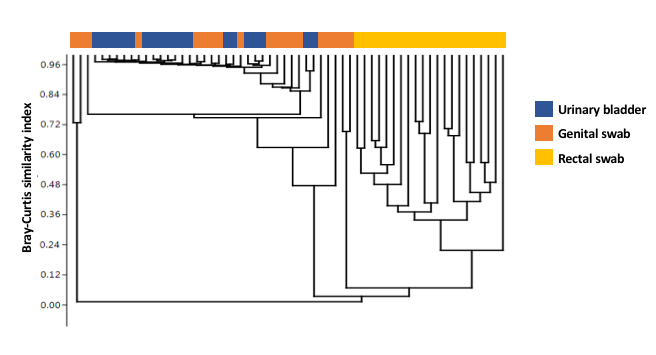
The urinary bladder in healthy dogs has dogmatically been considered free of bacteria. This study used culture independent techniques to characterize the healthy canine urinary microbiota. Urine samples collected by antepubic cystocentesis from dogs without urinary infection were used for DNA extraction. Genital tract and rectal samples were collected simultaneously from the same dogs. The V4 hypervariable region of the 16S rRNA bacterial gene was amplified and compared against Greengenes database for OTU assignment and relative abundance for urine, genital, and rectal samples. After excluding 4 dogs with cultivable bacteria, samples from 10 male (M; 1 intact) and 10 female (F) spayed dogs remained. All samples provided adequate genetic material for analysis. Four taxa (Pseudomonas sp., Acinetobacter sp., Sphingobium sp. and Bradyrhizobiaceae) dominated the urinary microbiota in all dogs of both sexes. These taxa were also detected in the genital swabs of both sexes, while the rectal microbiota differed substantially from the other sample sites. Principal component (PC) analysis of PC1 through PC3 showed overlap of urinary and genital microbiota and a clear separation of rectal swabs from the other sample sites along PC1, which explained 44.94% variation. Surprisingly, the urinary microbiota (mean # OTU 92.6 F, 90.2 M) was significantly richer than the genital (67.8 F, 66.6 M) or rectal microbiota (68.3 F, 71.2 M) (p < 0.0001), with no difference between sexes at any sample site. The canine urinary bladder is not a sterile environment and possesses its own unique and diverse microbiota compared to the rectal and genital microbiota. There was no difference between the sexes at any microbiota sample site (urine, genital, and rectal). The predominant bacterial genus for either sex in the urine and genital tracts was Pseudomonas sp.
See full study
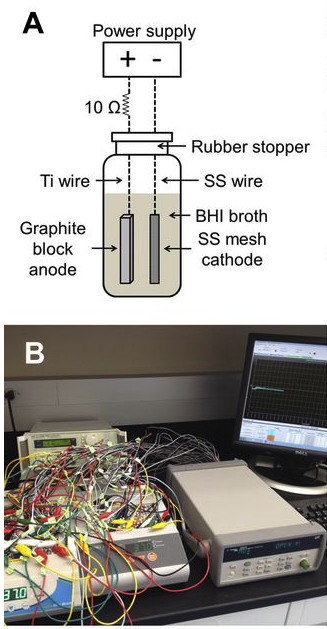
Ericsson, A.C., Davis, D.J., Franklin, C.L., and C.E. Hagan. (2015) Exoelectrogenic capacity of host microbiota predicts lymphocyte recruitment to the gut. Physiological Genomics, 47(7): 243-252. PMC4491528
See full study
Electrotaxis, directional cell movement in response to an electric potential, has been demonstrated in a wide range of cell types including lymphocytes. Exoelectrogens, microorganisms capable of generating electrical currents, have been identified in microbial fuel cells. However, no studies have investigated exoelectrogenic microbes in fresh feces, or the effects of an exoelectrogenic microbiota on the host organism. Here we show that commensal gut microbial populations differ in their capacity for electrical current production by exoelectrogens, and that those differences are predictive of increased lymphocyte trafficking to the gut in vivo, despite the lack of increased production of canonical lymphocyte-specific chemokines. Additionally, we demonstrate that the difference in current production between mice purchased from different commercial sources correlates reproducibly with the presence or absence of segmented filamentous bacteria, and while our data do not support a direct role for SFB in ex vivo current production, an exoelectrogenic microbiota can be transferred in vivo via mucosa-associated bacteria present in the ileum. Moreover, we detect upregulation of microbial genes associated with extracellular electron transfer in feces of mice colonized with exoelectrogenic microbiota containing segmented filamentous bacteria. While still correlative, these results suggest a novel means by which the gut microbiota modulates the recruitment of cells of the immune system to the gut.
Animal models provide a means whereby causal relationships between characteristic differences in the GM and diseases or conditions can be formally tested, using genetically identical animals in highly controlled environments. Clearly, the GM and its interactions with the host and myriad environmental factors are exceedingly complex and it is rare that a single microbial taxon associates with, much less causes, a phenotype with perfect sensitivity and specificity. Moreover, while the exact numbers are the subject of debate, it is well-recognized that only a minority of gut bacteria can be successfully cultured ex vivo. Thus, to perform studies investigating causal roles of the GM in animal model phenotypes, researchers need clever techniques to experimentally manipulate the GM of animals, and several ingenious methods of doing so have been developed, each providing its own type of information and with its own set of advantages and drawbacks. The current review will focus on the various means of experimentally manipulating the GM of research animals, drawing attention to the factors which would aid a researcher in selecting an experimental approach, and with an emphasis on mice and rats, the primary model species used to evaluate the contribution of the GM to a disease phenotype.
See full study
Segmented filamentous bacteria (SFB) modulate the ontogeny of the immune system, and their presence can significantly affect mouse models of disease. Until recently, the inability to culture SFB has made controlled studies of the mechanisms by which SFB exert their influence problematic. Herein, we demonstrate a means of selecting for SFB from a complex microbial mixture, providing researchers a simple and cost-effective means to prepare pure infective inocula for prospective studies, and compare individual isolates of SFB.
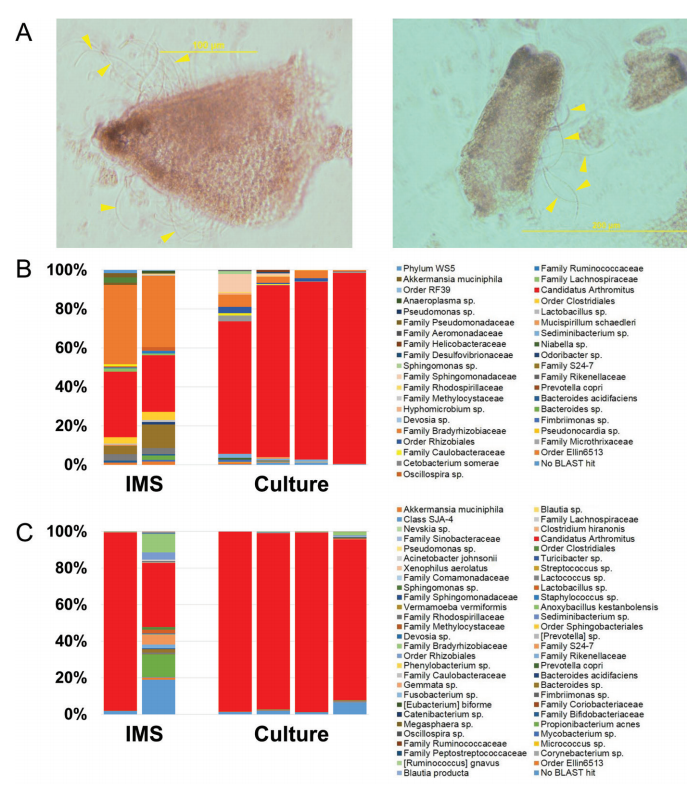
See full study
To evaluate the role of qualitatively different but naturally occurring gut microbiota and the relationship with colorectal cancer development, genetically identical embryos from the Polyposis in Rat Colon (Pirc) rat model of colorectal cancer were transferred into recipients of three different genetic backgrounds (F344/NHsd, LEW/SsNHsd, and Crl:SD). Our data show that the gut microbiota varies between rat strains, with LEW/SsNHsd having a greater relative abundance of the bacteria Prevotella copri. Both male and female F344-Pirc rats harboring the Lewis microbiota had decreased tumor burden relative to genetically identical rats harboring F344 or SD microbiota. Significant negative correlations were detected between tumor burden and the relative abundance of specific taxa from samples taken at weaning and shortly thereafter, prior to observable adenoma development. Notably, this naturally occurring variation in the gut microbiota is associated with a significant difference in severity of colorectal cancer, and the abundance of certain taxa is associated with decreased tumor burden.
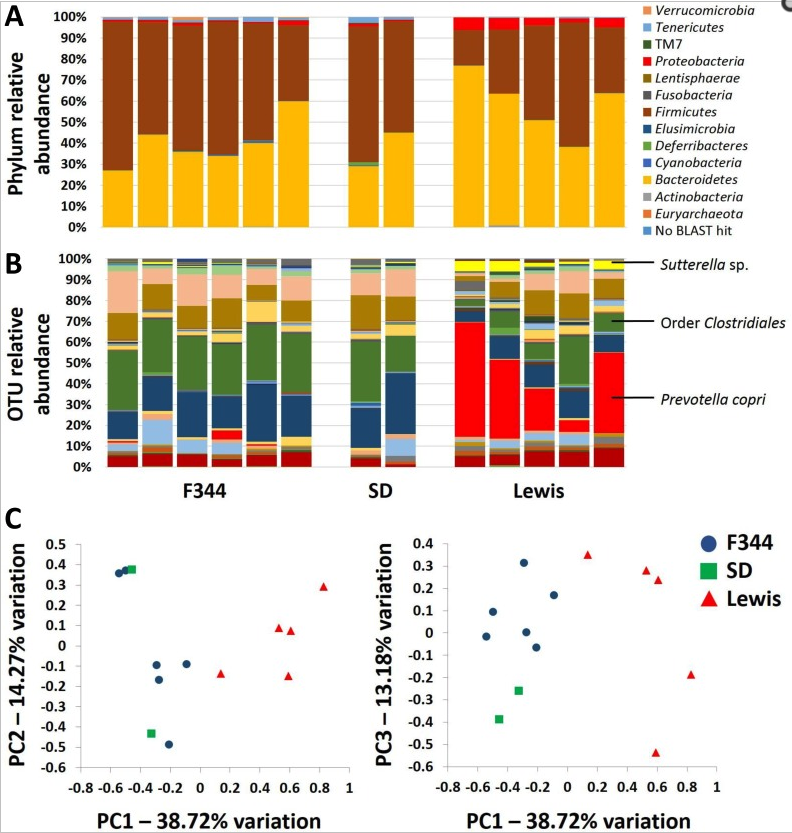
Ericsson, A.C., Akter, S., Hanson, M., Busi, S.B., Parker, T., Schehr, R., Hankins, M., Ahner, C.E., Davis, J.W., Franklin, C.L., Amos-Landgraf, J., and E.C. Bryda. (2015) Differential susceptibility to colorectal cancer due to naturally occurring gut microbiota. Oncotarget, 6(32):33689-33704. PMID:26378041 (PMCID in process)
See full study
To determine the influence of extraction technique on the success of PCA amplification and 16S RNA amplicon sequencing, we investigated the use of five common DNA extraction methods on fecal samples from five different species. Our data show that the method of DNA extraction impacts DNA concentration and purity and successful NGS amplification, and influences microbial communities detected in sequencing output dependent on the species of fecal sample and the DNA extraction method used. These data highlight the importance of careful consideration of DNA extraction method when designing and interpreting data from cross species studies.
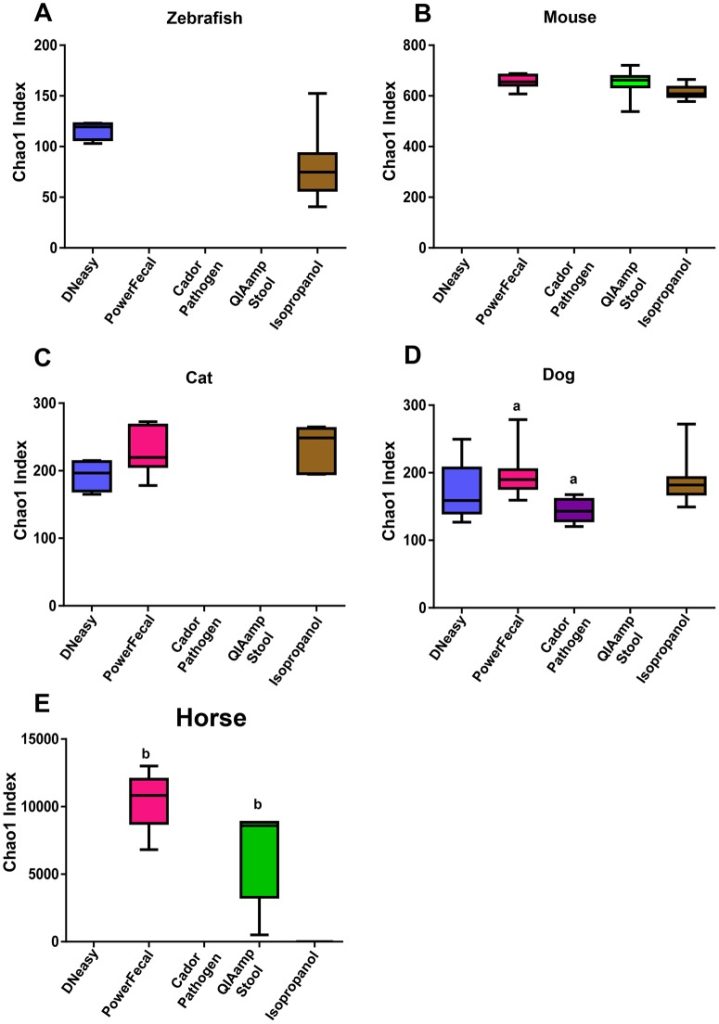
See full study
Tumor necrosis factor alpha (TNF) antagonists are beneficial to patients with inflammatory autoimmune diseases, including Crohn’s disease, rheumatoid arthritis, and ankylosing spondylitis. Paradoxically, anti-TNF therapeutics exacerbate CNS demyelinating autoimmune diseases such as multiple sclerosis (MS), neuromyelitis optica (NMO), and optic neuritis (ON). The underlying mechanisms remain enigmatic. Here, we demonstrate that genetic deficiency of TNF receptor type 2 (TNFR2) in myelin oligodendrocyte glycoprotein (MOG) 35-55–specific TCR (2D2) transgenic mice results in a highly penetrant female-biased spontaneous inflammatory demyelination that histologically resembles autoimmune NMO, also called Devic’s disease. Antibiotic treatment resulted in almost complete abrogation of clinical signs in female mice, and sequencing the V4 region of fecal bacterial 16S rRNA of TNFR2-/- 2D2 and 2D2 mice revealed overlapping, but distinct, gut bacterial communities that associated with sexed-bias autoimmune disease susceptibility.
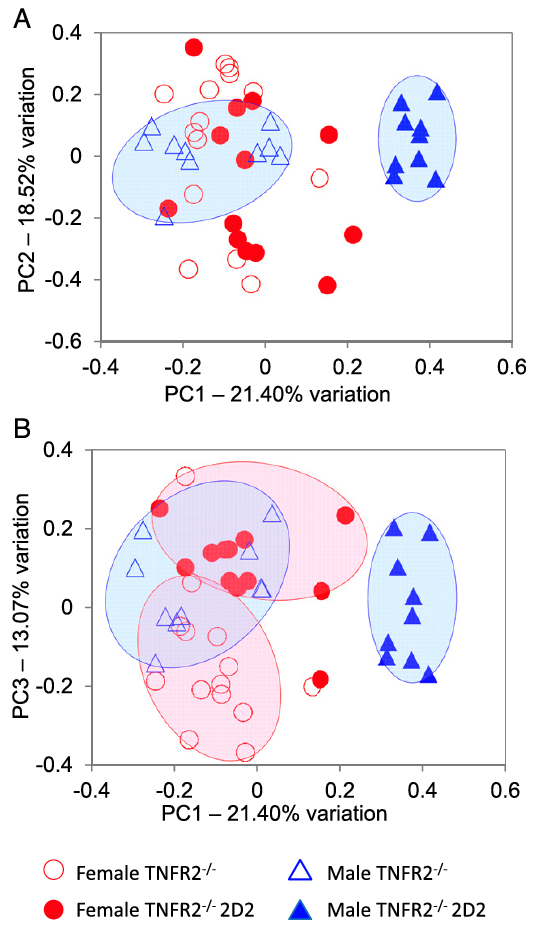
Miller, P.G., Bonn, M.B., Franklin, C.L., Ericsson, A.C., and S.C. McKarns. (2015) TNFR2 Deficiency Acts in Concert with Gut Microbiota to Precipitate Sex-biased Spontaneous CNS Demyelinating Autoimmune Disease. Journal of Immunology, 195(10):4668-4684. PMID:26475926 (PMCID in process)
See full study
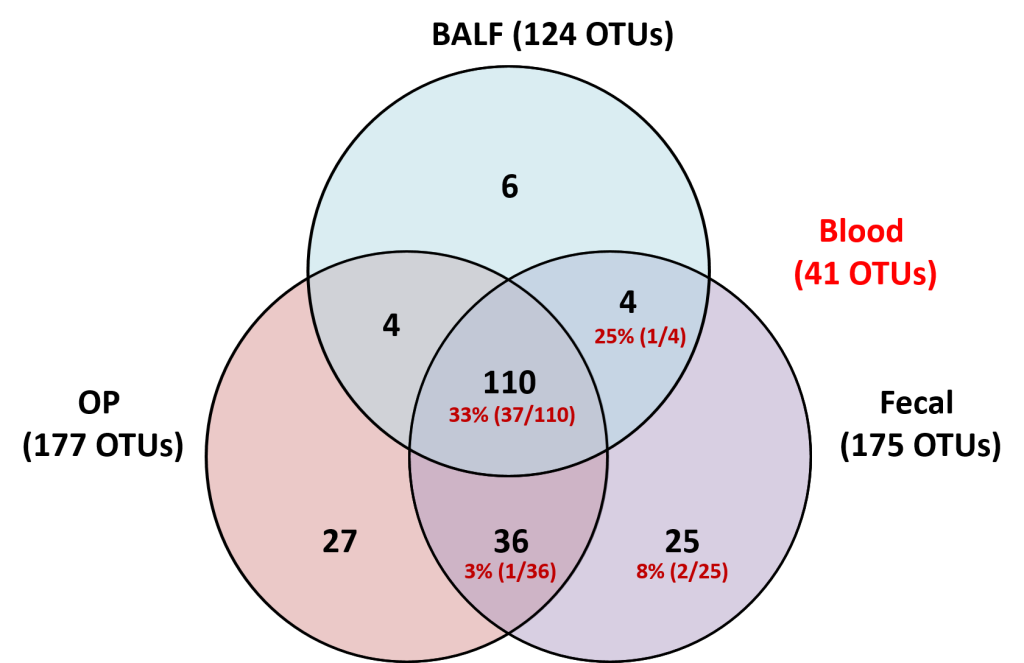
The upper and lower airways of healthy humans are reported to harbor stable and consistent bacterial populations, and the composition of these communities is altered in individuals affected with several respiratory diseases. Data regarding the presence of airway microbiota in other animals are scant and a better understanding of the composition and metabolic function of such bacterial populations is essential for the development of novel therapeutic and diagnostic modalities for use in both veterinary and human medicine. Based on targeted next-generation sequencing of feces and samples collected at multiple levels of the airways from 16 healthy female dogs, we demonstrate that canine airways harbor a topographically continuous microbiota with increasing relative abundance of proteobacterial species from the upper to lower airways. The lung-associated microbiota, as assessed via bronchoalveolar lavage fluid (BALF), was the most consistent between dogs and was dominated by three distinct taxa, two of which were resolved to the species level and one to the level of family. The gene content of the nasal, oropharyngeal, and lung-associated microbiota, predicted using the Phylogenetic Investigations into Communities by Reconstruction of Unobserved States (PICRUSt) software, provided information regarding the glyoxylate and citrate cycle metabolic pathways utilized by these bacterial populations to colonize such nutrient-poor, low-throughput environments. These data generated in healthy subjects provide context for future analysis of diseased canine airways. Moreover, as dogs have similar respiratory anatomy, physiology, and immune systems as humans, are exposed to many of the same environmental stimuli, and spontaneously develop similar respiratory diseases, these data support the use of dogs as a model species for prospective studies of the airway microbiota, with findings translatable to the human condition.
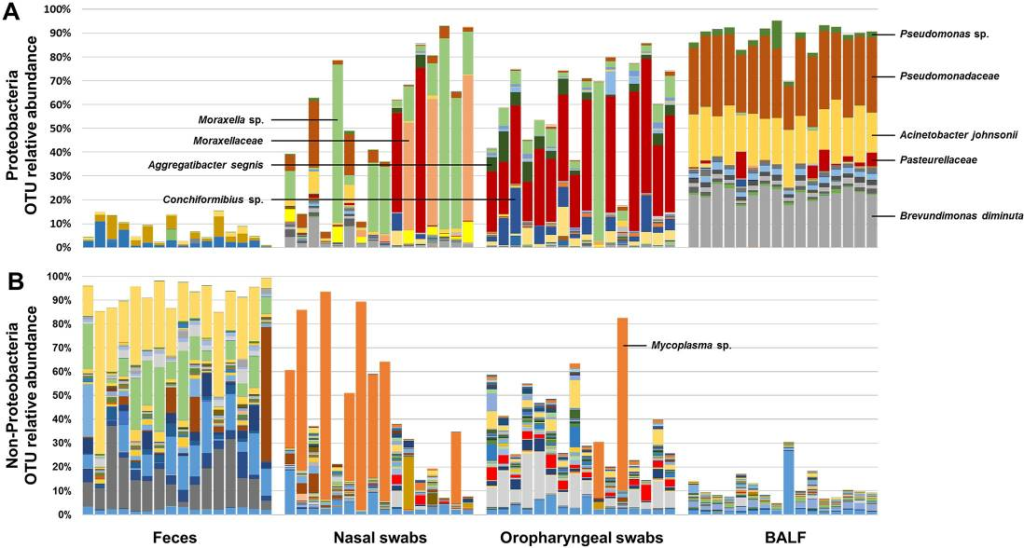
See full study
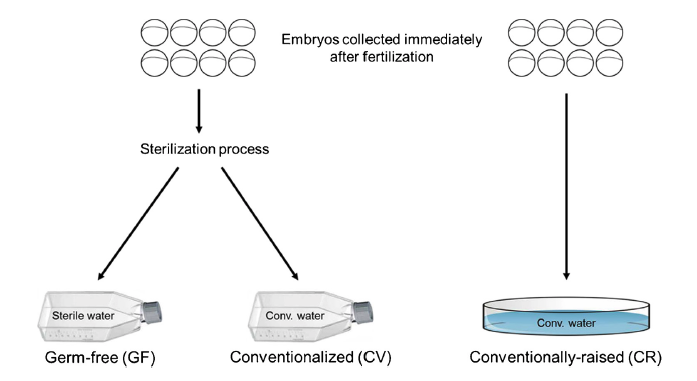
Data presented here contains metagenomic analysis regarding the sequential conventionalization of germ-free zebrafish embryos. Zebrafish embryos that underwent a germ-free sterilization process immediately after fertilization were promptly exposed to and raised to larval stage in conventional fish water. At 6 days post fertilization (dpf), these “conventionalized” larvae were compared to zebrafish larvae that were raised in conventional fish water never undergoing the initial sterilization process. Bacterial 16S rRNA amplicon sequencing was performed on DNA isolated from homogenates of the larvae revealing distinct microbiota variations between the two groups. The dataset described here is also related to the research article entitled “Microbial modulation of behavior and stress responses in zebrafish larvae”.
Davis, D.J., Bryda, E.C., Gillespie, C.H., and A.C. Ericsson. (2016) 16S rRNA amplicon sequencing dataset for conventionalized and conventionally raised zebrafish larvae. Data in Brief, 8:938-943. PMC4961299
See full study
The influence of the microbiota on behavior and stress responses is poorly understood. Zebrafish larvae have unique characteristics that are advantageous for neuroimmune research, however, they are currently underutilized for such studies. Here, we used germ-free zebrafish to determine the effects of the microbiota on behavior and stress testing. The absence of a microbiota dramatically altered locomotor and anxiety-related behavior. Additionally, characteristic responses to an acute stressor were also obliterated in larvae lacking exposure to microbes. Moreover, these phenomena were not able to be reverted by exposure to exogenous bacterial metabolites introduced via immersion. Lastly, treatment with the probiotic Lactobacillus plantarum was sufficient to attenuate anxiety-related behavior in conventionally-raised zebrafish larvae. These results underscore the importance of the microbiota in communicating to the CNS via the microbiome-gut-brain axis and set a foundation for using zebrafish larvae for neuroimmune research.
See full study
Enterotoxigenic Escherichia coli (ETEC) causes diarrheal disease. Antigenic and structural heterogeneity among ETEC colonization factors has complicated vaccine development efforts. Identifying and characterizing conserved ETEC antigens that induce protective immunity is therefore of interest. We previously characterized three proteins (MipA, Skp, and ETEC_2479) that protected mice in an intranasal ETEC challenge model after vaccination. However, these proteins are conserved not only in multiple ETEC isolates, but also in commensal bacteria. While the impact of inactivated viral vaccines and live-attenuated bacterial vaccines on the host microbiota have been examined, the potential impact of using subunit vaccines consisting of antigens that are also encoded by commensal organisms has not been investigated. We addressed this issue by characterizing changes to mouse intestinal microbiomes as a function of vaccination. We failed to observe significant changes to mouse health, to mouse weight gain as a function of time, or to the diversity or richness of mouse intestinal microbiomes, as measured by analyzing alpha- and beta-diversity, as well as overall community structure, before and after vaccination.
Hays, M.P., Ericsson, A.C., Yang, Y., and P.R. Hardwidge. (2016) Vaccinating with conserved E. coli antigens does not alter the mouse intestinal microbiome. BMC Research Notes, 9:401-407. PMC4981990
See full study
Puppies in a research setting frequently have diarrheal disease just prior to weaning or at weaning (approximately 8 to 10 weeks of age). This may be due, in part, to an immature and unstable intestinal microbiota that is permissive to an opportunistic pathogen such as a Coccidian species. The overall objective of this study was to assess whether fecal microbiota transfer (FMT) would result in more rapid transmission of stable maternal microbiota to pups and a lower frequency of post-weaning diarrhea. Puppies were designated by litter as treated (FMT) or sham-treated. The FMT group was administered fecal inoculum for 5 consecutive days during weaning. Diarrhea in both litters was evaluated using the Nestlé Purina Fecal Scoring System for 10 days during the weaning period. Fresh feces were collected from dams and puppies at 3 days prior to weaning and 3, 10, and 24 days post weaning for analysis of the fecal microbiota using 16S rRNA amplicon sequencing. The composition of fecal inoculum refrigerated at temperatures between 3 and 5°C was stable for at least 5 days. No diarrhea was reported in either group during the study period, making comparison of treated and control groups problematic. 16S rRNA gene analysis, however, did reveal microbial variability across time in both. Thus, while neither group appeared to mirror the dam at any of the designated time points during the study, the data provided fundamental and novel information regarding the dynamic instability of the fecal microbiota of puppies following weaning.
Burton, E., O’Connor, E., Ericsson, A.C., and C.L. Franklin. (2016) Evaluation of fecal microbiota transfer as treatment for post-weaning diarrhea in research-colony puppies. Journal of the American Association for Laboratory Animal Science, 55(5):582-587. PMID:27657714 (PMCID in process)
See full study
The consumption of probiotics has become increasingly popular as a means to try to improve health and well-being. Not only are probiotics considered beneficial to digestive health, but increasing evidence suggests direct and indirect interactions between gut microbiota (GM) and the central nervous system (CNS). Here, adult zebrafish were supplemented with Lactobacillus plantarum to determine the effects of probiotic treatment on structural and functional changes of the GM, as well as host neurological and behavioral changes. L. plantarum administration altered the β-diversity of the GM while leaving the major core architecture intact. These minor structural changes were accompanied by significant enrichment of several predicted metabolic pathways. In addition to GM modifications, L. plantarum treatment also significantly reduced anxiety-related behavior and altered GABAergic and serotonergic signaling in the brain. Lastly, L. plantarum supplementation provided protection against stress-induced dysbiosis of the GM. These results underscore the influence commensal microbes have on physiological function in the host, and demonstrate bidirectional communication between the GM and the host.
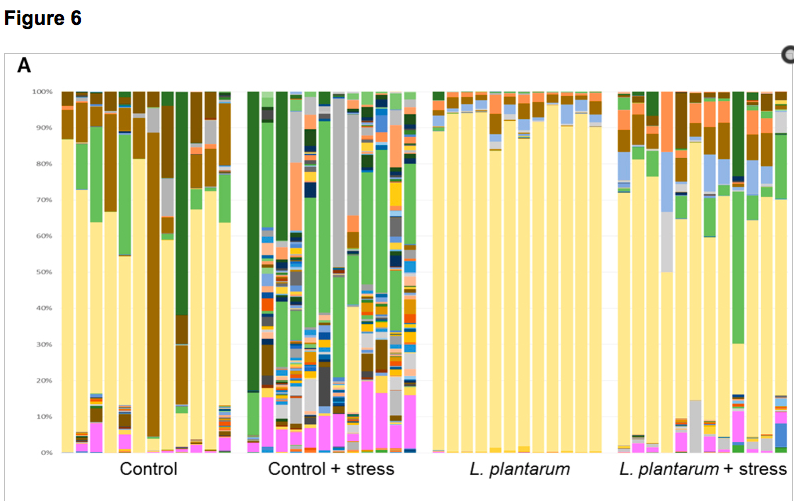
See full study
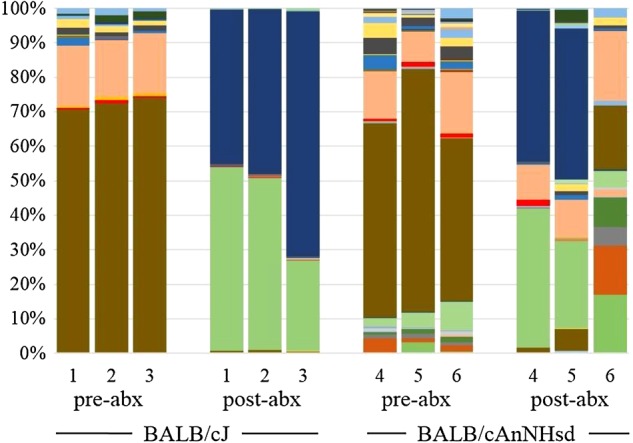
There is abundant evidence that the gut microbiota of inbred laboratory mice may modulate the phenotype of disease models. It is recognized that the gut microbiota of mice differs dependent on genetic background and commercial source of mice. Using culture-independent methods, we demonstrate significant differences in the richness, diversity, and composition of fecal microbiota in mice purchased from two large commercial vendors. These data provide the first in-depth analysis of the developmental trajectory of the fecal microbiota in mice from different vendors, and a starting point from which researchers may be able to refine animal models affected by differences in the gut microbiota.
Segmented filamentous bacteria (SFB) represent long-recognized commensal bacteria capable of modulating mouse models of disease, due largely to effects on the ontogeny of the adaptive immune system. Herein we review the basic biology of SFB and their impact on mouse models of disease.